
Details of the Drug
General Information of Drug (ID: DM9SVEB)
| Drug Name |
4-Chloro-7-methyl-2-phenyl-[1,8]naphthyridine
|
||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Synonyms | CHEMBL97778; 4-chloro-7-methyl-2-phenyl-1,8-naphthyridine; 4-Chloro-7-methyl-2-phenyl-[1,8]naphthyridine; SCHEMBL1536334; BDBM50090689; ZINC13579501 | ||||||||||||||||||||||
| Indication |
|
||||||||||||||||||||||
| Drug Type |
Small molecular drug
|
||||||||||||||||||||||
| Structure |
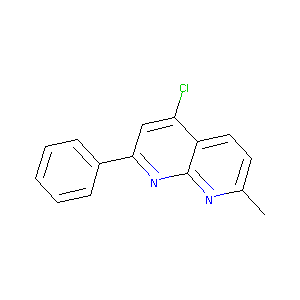 |
||||||||||||||||||||||
| 3D MOL | 2D MOL | ||||||||||||||||||||||
| #Ro5 Violations (Lipinski): 0 | Molecular Weight (mw) | 254.71 | |||||||||||||||||||||
| Logarithm of the Partition Coefficient (xlogp) | 4.1 | ||||||||||||||||||||||
| Rotatable Bond Count (rotbonds) | 1 | ||||||||||||||||||||||
| Hydrogen Bond Donor Count (hbonddonor) | 0 | ||||||||||||||||||||||
| Hydrogen Bond Acceptor Count (hbondacc) | 2 | ||||||||||||||||||||||
| Chemical Identifiers |
|
||||||||||||||||||||||
| Cross-matching ID | |||||||||||||||||||||||
Molecular Interaction Atlas of This Drug
Drug Therapeutic Target (DTT) |
|
||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Molecular Interaction Atlas (MIA) | |||||||||||||||||||||||||||